


-
Book Overview & Buying

-
Table Of Contents

Mastering Machine Learning Algorithms - Second Edition
By :
Mastering Machine Learning Algorithms
By:
Overview of this book
Mastering Machine Learning Algorithms, Second Edition helps you harness the real power of machine learning algorithms in order to implement smarter ways of meeting today's overwhelming data needs. This newly updated and revised guide will help you master algorithms used widely in semi-supervised learning, reinforcement learning, supervised learning, and unsupervised learning domains.
You will use all the modern libraries from the Python ecosystem – including NumPy and Keras – to extract features from varied complexities of data. Ranging from Bayesian models to the Markov chain Monte Carlo algorithm to Hidden Markov models, this machine learning book teaches you how to extract features from your dataset, perform complex dimensionality reduction, and train supervised and semi-supervised models by making use of Python-based libraries such as scikit-learn. You will also discover practical applications for complex techniques such as maximum likelihood estimation, Hebbian learning, and ensemble learning, and how to use TensorFlow 2.x to train effective deep neural networks.
By the end of this book, you will be ready to implement and solve end-to-end machine learning problems and use case scenarios.
Table of Contents (28 chapters)
Preface

Machine Learning Model Fundamentals
 Free Chapter
Free Chapter
Loss Functions and Regularization
Introduction to Semi-Supervised Learning
Advanced Semi-Supervised Classification
Graph-Based Semi-Supervised Learning
Clustering and Unsupervised Models
Advanced Clustering and Unsupervised Models
Clustering and Unsupervised Models for Marketing
Generalized Linear Models and Regression
Introduction to Time-Series Analysis
Bayesian Networks and Hidden Markov Models
The EM Algorithm
Component Analysis and Dimensionality Reduction
Hebbian Learning
Fundamentals of Ensemble Learning
Advanced Boosting Algorithms
Modeling Neural Networks
Optimizing Neural Networks
Deep Convolutional Networks
Recurrent Neural Networks
Autoencoders
Introduction to Generative Adversarial Networks
Deep Belief Networks
Introduction to Reinforcement Learning
Advanced Policy Estimation Algorithms
Other Books You May Enjoy
Index

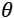 and its relationship with the real world
and its relationship with the real world






